



| 台灣每年約有 3/1000 左右的新生兒患有「感音神經性聽力損失」,其中2/1000是因父母的GJB2、SLC26A4 及粒線體12S rRNA等突變基因遺傳所造成的遺傳性聽力損失。語前聽力損失對幼兒的學習成長會有非常大的影響,盡早了解是否有聽損基因的相關遺傳風險是非常必要的。本篇文章將帶您了解聽力損失之成因及治療方式,以及該如何預防下一代患病。 |
| 文章目錄: 一、聽力損失是什麼? 帶您認識遺傳性/非遺傳性聽力損失 二、GJB2 基因有什麼作用?是如何遺傳的? 三、GJB2 基因突變率高嗎? 突變可能造成什麼疾病? 四、SLC26A4 基因有什麼作用?是如何遺傳的? 五、SLC26A4 基因突變率高嗎? 突變可能造成什麼疾病? 六、遺傳性聽損可以避免嗎?孩子遭遇聽力損失怎麼辦? 七、目前有哪些聽力檢測或治療的方式嗎? |
聲音是經由外耳進入耳道、引起鼓膜震動,中耳聽小骨再將震動傳遞給毛細胞,將震動轉化為電脈衝後,藉由內耳聽覺神經傳遞訊號給大腦,讓我們能分辨語音、樂音,以聲音感受這個世界。
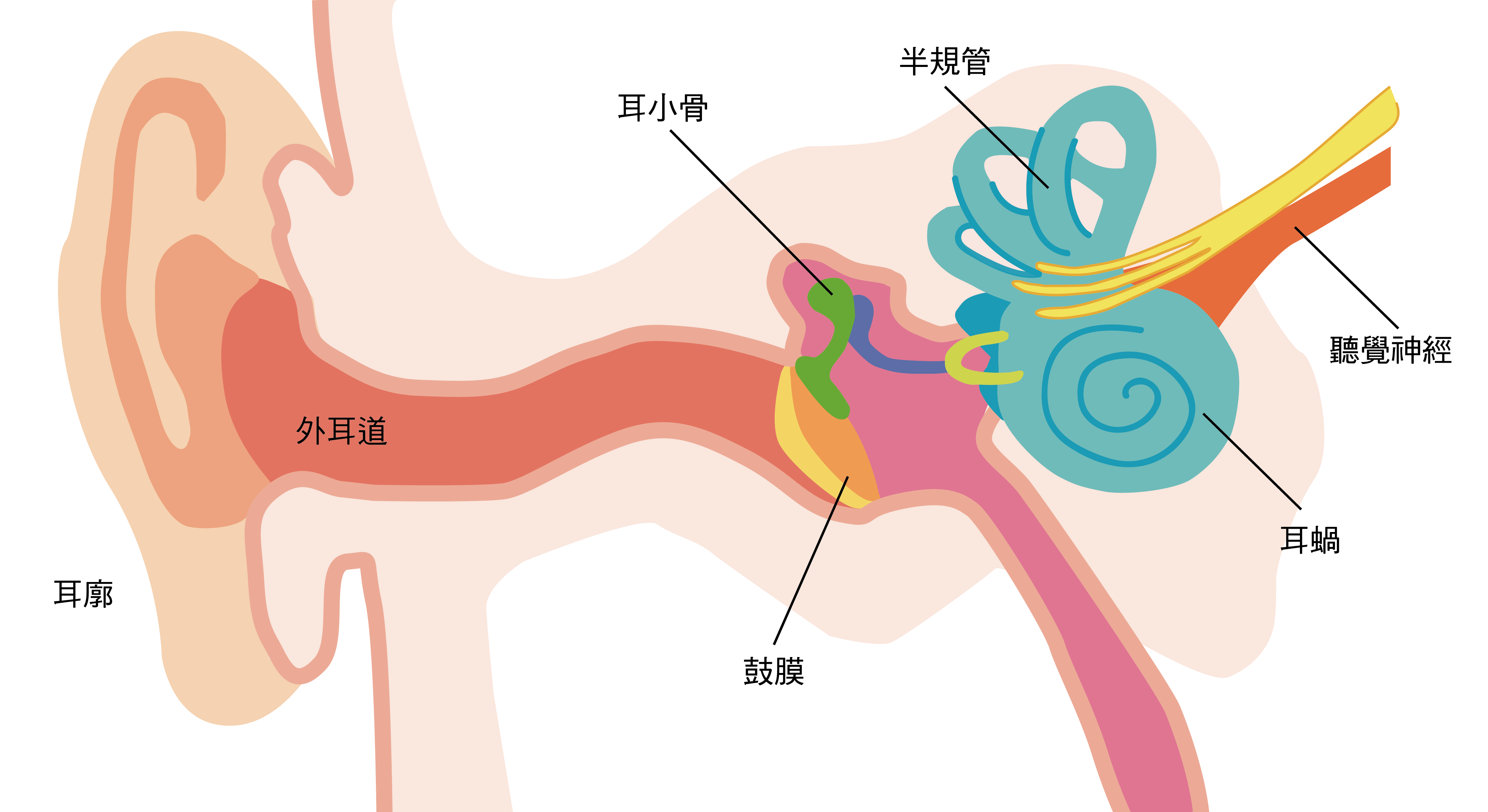
聽力損失就是聲音由外耳→中耳→內耳依序傳導的流程出現問題,可依發生時間區分為「先天型聽損」及「後天型聽損」。先天型聽損是胎兒在懷孕期間或出生時因病毒感染等原因導致,後天型聽損則指出生後因疾病、外傷、老化等各種因素導致聽損。
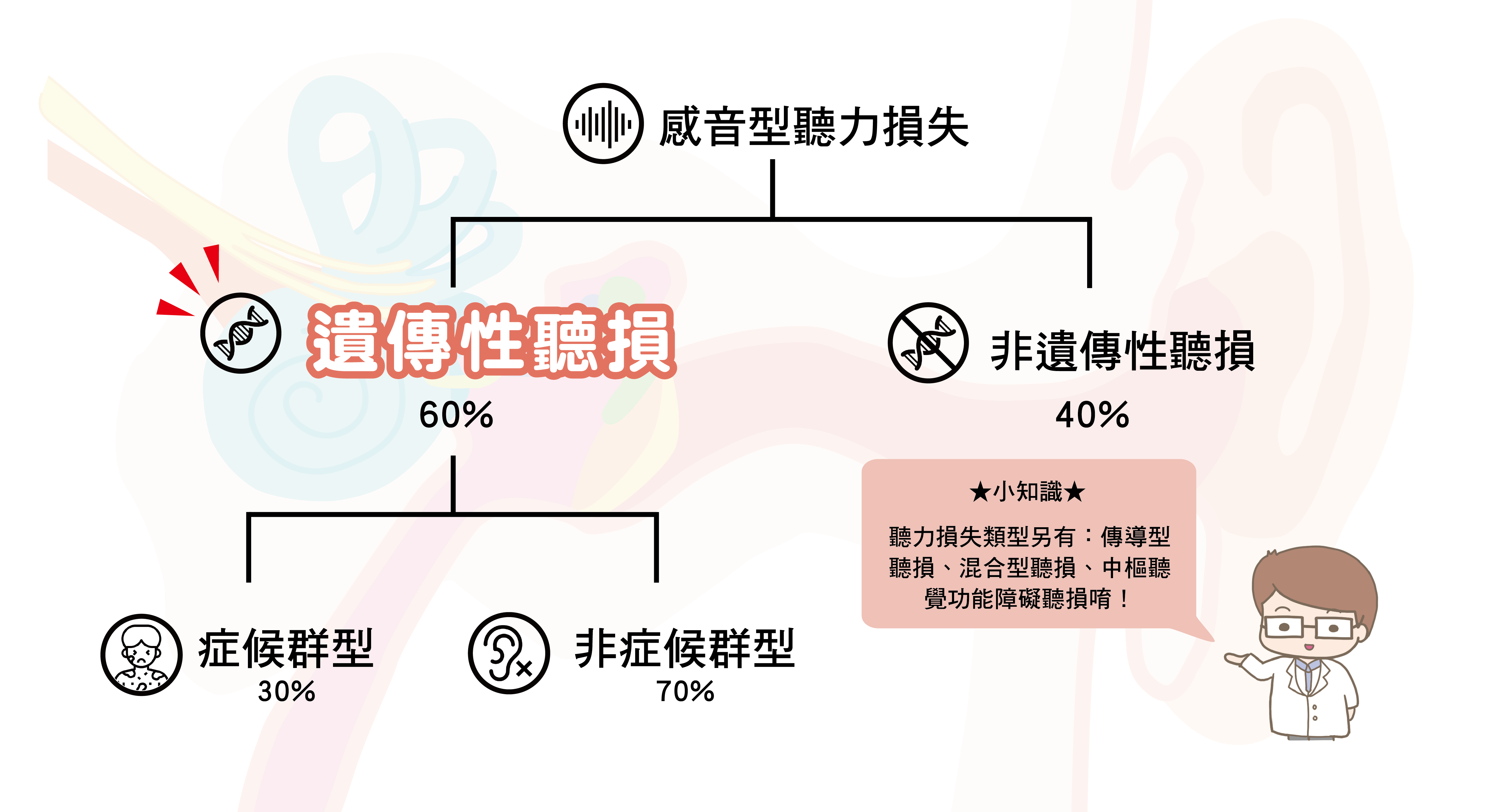
依據病因【聽力損失】在醫學上可分為以下4個類型:
| 聽力損失類型 | 器官 | 說明 |
| 傳導性聽損 | 外耳、中耳 | 耳聽小骨的異常 |
| 感音神經性聽損 | 內耳 | 結構異常(包含耳蝸及聽覺神經等) |
| 混合型聽損 | 綜合 | 傳導型聽力損失及感音神經性聽力損失同時發生 |
| 中樞聽覺功能障礙 | 顱內聽覺器官 | 腦部神經或器官發生受損或功能障礙 |
台灣每年約有 3/1000 左右的新生兒患有「感音神經性聽力損失」,其中有2/1000左右是基因突變造成的遺傳性聽力損失,另外 1/1000 則是由子宮內感染(梅毒或巨細胞病毒)、腦炎、早產等影響所導致的非遺傳性聽力損失。
在台灣遺傳性聽力損失的患者中,最常觀察到突變的基因有GJB2、SLC26A4 及粒線體12S rRNA等。大約30%患者除聽力損失外,體內其他器官也有病變出現,稱為症候群型遺傳性聽力損失,像是 Pendred氏症候群、Usher症候群等。另外70%患者僅有聽力損失症狀、沒有其他病變,稱為非症候群型遺傳性聽力損失。
GJB2 基因有什麼作用?是如何遺傳的?
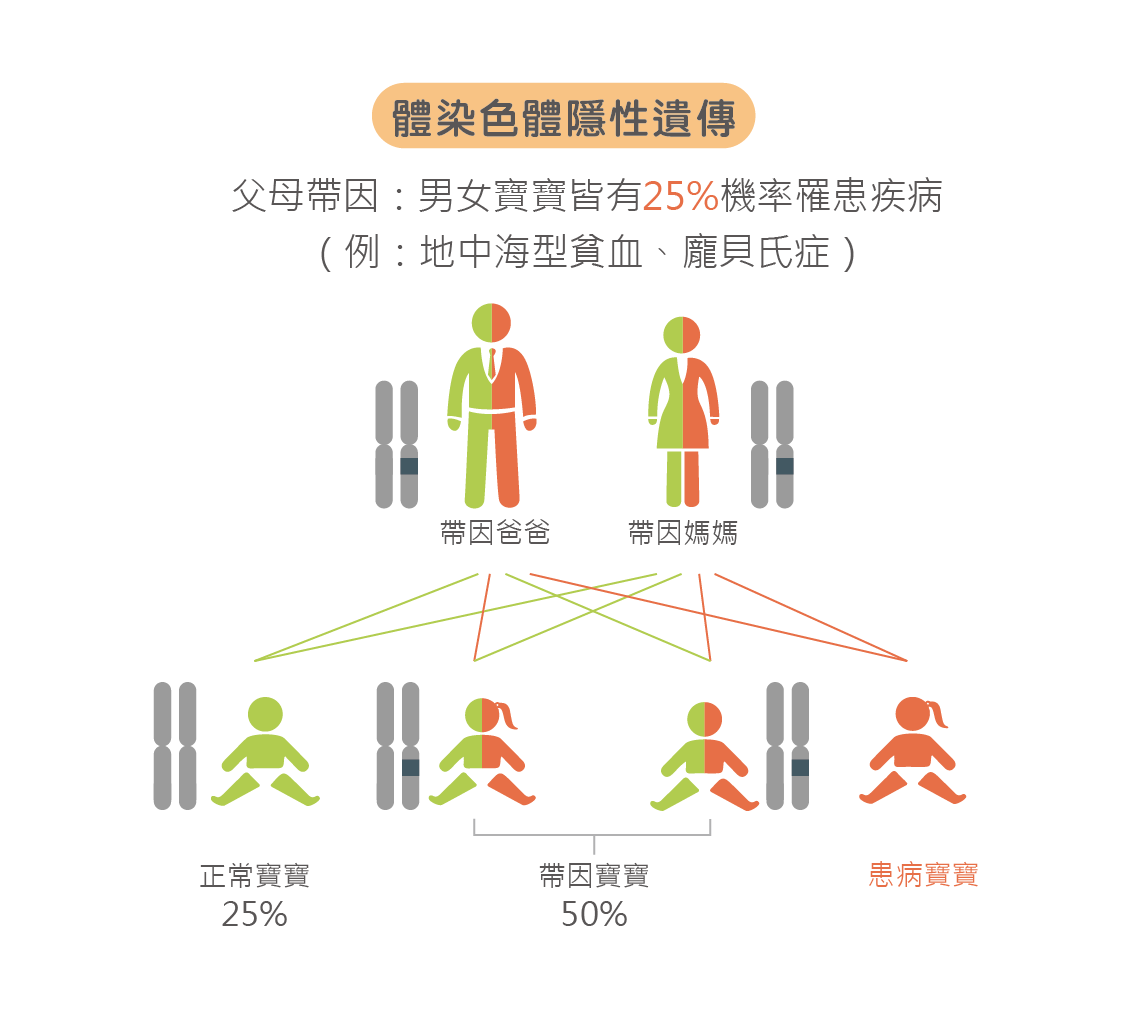
GJB2 基因位在第 13 號染色體 q12.11位置,能夠表現間隙接合蛋白 26 (Connexin 26),其功能為維持內耳淋巴液的離子濃度穩定,使聲音的震動能轉換為電脈衝傳導給聽神經;當此基因發生突變時,會觀察到患者的聽力受損,但不會引起其他部位病變。 GJB2 基因異常是導致非症候群遺傳性聽力損失的主要突變基因之一,其遺傳模式為體染色體隱性遺傳,也就是即使雙親皆無遺傳性聽力損失問題,若正好皆為帶因者,胎兒就有25%的機會發病。
GJB2 基因突變率高嗎? 突變可能造成什麼疾病?
世界衛生組織統計發現目前全球約有 5%左右人口有耳聾或聽力損失等相關問題(包含 4.3 億名成人及 3400 萬名兒童);在台灣近年研究中,約17.3%的聽力損失患者被檢查出 GJB2 基因有突變的狀況,常見的突變位點有c.109G>A (p.V37I;85.9%)、c.235delC (11.5%)及c.299_300delAT (1.7%)等,GJB2基因可能導致語前中、重度聽力損失,或有隨年齡逐漸惡化的聽力損失症狀。此外,語前聽力損失對幼兒的學習成長會有非常大的影響,盡早了解是否有聽損基因的相關遺傳風險是非常必要的。
SLC26A4 基因有什麼作用?是如何遺傳的?
SLC26A4 基因位在第7號染色體 q22.3位置,此基因轉譯出陰離子運輸蛋白 Pendrin,負責調節碳酸氫根(HCO3-)及氯離子(Cl-)以維持內耳淋巴液的離子濃度穩定,使聲音的震動能轉換為電脈衝傳導給聽神經;此外,Pendrin在甲狀腺中也負責調節碘離子(I-)濃度,故 SLC26A4 基因也是症候群型聽力損失 - Pendred氏症候群的主要致病基因,此突變導致患者有前庭導水管增大(Enlarged Vestibular Aqueduct, EVA)、Mondini氏發育不全(Mondini’s dysplasia)及甲狀腺腫大等症狀,因此 SLC26A4 基因又稱 PDS 基因。
SLC26A4 基因異常可能導致症候群或非症候群遺傳性聽力損失,其遺傳模式為體染色體隱性遺傳,也就是即使雙親皆無遺傳性聽力損失問題,若正好皆為帶因者,胎兒就有25%的機會發病。
SLC26A4 基因突變率高嗎? 突變可能造成什麼疾病?
在台灣近年研究中,約 6.03%的聽力損失患者被檢查出 SLC26A4 基因有突變的狀況,其中有83.1%患者為雙對偶基因突變(bi-allelic,兩條染色體的基因都突變),12.7%患者為單對偶基因突變(mono-allelic);常見的突變位點有c.919-2A>G(80%)、c.2168A >G(6%)及 c.1229C >T(2.6%)等。
SLC26A4 基因突變可能導致內耳構造畸形及波動型聽力喪失症狀,內耳構造畸形包含前庭導水管擴大及Mondini氏發育不全,會導致語前中重度聽力損失;波動型聽力喪失則指患者聽力時好時壞,當受到外力影響時(如頭部撞擊)甚至可能會發生聽力永久損失的狀況。值得注意的是,語前聽力損失對幼兒的學習成長會有非常大的影響,盡早了解是否有聽損基因的相關遺傳風險是非常必要的。
遺傳性聽損可以避免嗎?孩子遭遇聽力損失怎麼辦?
遺傳性聽力損失可能導致孩子語言發展遲緩並產生學習障礙,不僅孩子將承受諸多不便與辛苦,爸爸媽媽們也會面臨龐大的心理負擔及照護壓力,對此我們感同身受,希望能最大程度降低遺傳性聽損的發生率。現今單基因遺傳疾病基因檢測,利用次世代基因定序(NGS)技術,一次試驗即可篩檢一百多項單基因隱性遺傳項目;除了 GJB2 基因相關遺傳性聽力損失,還涵蓋了身體內/外器官之異常疾病(如:肌肉萎縮症、白化症、魚鱗癬病等),呼吸、循環、代謝系統異常疾病(如:地中海型貧血、血友病等),一次檢測、終身受用。
建議夫妻雙方可在婚前健檢、備孕階段或懷孕初期就提早篩檢,了解自己是否有疾病帶因的風險,針對雙親均帶因的異常疾病,盡早與醫師規劃胎兒的相關檢查。

目前有哪些聽力檢測或治療的方式嗎?
國內目前有針對新生兒聽力檢測的補助(合約院所請點此,將開新視窗前往),通常會在出生後24小時至出院、滿月前完成初篩或複篩,施作項目為「自動型聽性腦幹反應」(aABR),其原理為利用耳機提供聲音刺激後、經由貼在頭部皮膚表層的電極接收來自耳蝸、聽神經及腦幹所傳出的神經電位反應,不易受新生兒外耳道胎脂影響而造成偽陽性反應;若不幸未通過聽力篩檢,需再由醫療機構做診斷檢查(合約院所請點此,將開新視窗前往),包含電生理檢查(如:聽性腦幹反應、耳聲傳射檢查、中耳功能檢查、穩定狀態誘發聽覺電位反應等),及行為檢查(如:聽力行為觀察、視覺回饋聽力檢查等),經醫師評估後可能也會進一步進行遺傳基因檢查或電腦斷層(CT)、磁振照影(MRI),以確認聽力損失的原因、程度及種類。聽力損失的治療方式包含有聽力輔具的使用 (如:助聽器、人工電子耳植入)、口語及手語的聽能復健教學等,只要能夠與醫師詳細諮詢溝通,早期介入治療,就能盡可能減少聽力損失對孩子各方面發展的影響。
參考資料:
1. Chen-Chi Wu, et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes. 2019.
2. A Eliot Shearer, et al. Hereditary Hearing Loss and Deafness Overview. GeneReviews® 2017.
3. 台灣新生兒聽力篩檢與確診指引手冊 (2014)
|
|
.png)
撰文 / Rex Liu
校閱 / Zong Ye , Jora Li
文章最後更新時間:2022/09/16
(#台灣基康145)


