



| 脊髓性肌肉萎縮症(SMA)是體染色體最常見帶因的單基因遺傳病,主要影響嬰兒及幼童族群。患者肌肉會萎縮無力至無法自理生活,甚至因其他併發症而影響壽命。如適齡夫妻準備懷孕或是正值懷孕初期,建議施作帶因者篩檢以及早檢測有無攜帶致病基因,了解自身基因是否有遺傳的風險。本篇文章將帶您了解導致脊髓性肌肉萎縮症的致病原因及可施作哪些檢查項目。 |
| 文章目錄: 1. 什麼是脊髓性肌肉萎縮症(SMA)? 2. 為什麼會導致脊髓性肌肉萎縮症(SMA)? 3. 脊髓性肌肉萎縮症(SMA)有不同的嚴重程度? 4. 脊髓性肌肉萎縮症是怎麼遺傳? 5. 脊髓性肌肉萎縮症(SMA)的治療與照護 6. 孕前或懷孕初期儘早做SMA基因篩檢,守護寶寶健康 |
什麼是脊髓性肌肉萎縮症(SMA)?
脊髓性肌肉萎縮症(Spinal Muscular Atrophy, 簡稱SMA)為一種神經肌肉退化疾病的遺傳疾病, SMA除了是導致全球嬰兒死亡率最高的遺傳疾病,也是台灣常見的單基因遺傳病。根據統計,SMA患者佔約一萬分之一的全球人口1,而一項2005年至2009年的統計數據更顯示,每5,000~10,000位台灣新生兒中就約有1位飽受患病之苦2。
常見症狀
SMA患者因為脊髓的前角運動神經元出現漸進式退化,進而造成肌肉出現萎縮無力,主要影響患者的肌肉如肩膀、大腿以及骨盆等部位。患者無法自主控制頭部運動,且會失去坐立、走路等運動能力,隨著年齡增長,患者甚至會出現呼吸及吞嚥困難等症狀。
智力發展
雖然每個患者的肌肉退化程度隨年齡及分型會有所不同,但此症並不影響患者的智力與認知發展。

▲脊髓性肌肉萎縮症(SMA)的典型症狀。▲
為什麼會導致脊髓性肌肉萎縮症(SMA)?
脊髓性肌肉萎縮症是因位於第 5 號染色體的 SMN1 基因缺失或突變所導致的遺傳疾病。SMN 基因主要負責製造運動神經元存活(Survival motor neuron,SMN)蛋白,作為大腦與肌肉之間傳遞訊息的重要橋樑。若 SMN1 基因產生突變,患者的肌肉無法接收到來自大腦的訊號,後期因肌細胞無法正常收縮,造成逐漸退化至喪失功能,進而導致全身肌肉的萎縮,約有95% SMA 患者主要成因為 SMN1 基因異常造成。
一般人在第5號染色體上會帶有兩種SMN 基因,分別為 SMN1 及 SMN2 基因。兩者皆能製造 SMN 蛋白,差異在於所製造的 SMN 蛋白濃度與功能3:
-
SMN1基因:為 SMN 蛋白的主要製作者,能製造大量且具正常功能的 SMN 蛋白;
-
SMN2基因:大多生成為結構較不穩定 SMN 蛋白,僅能產生較少量的正常 SMN,因此無法維持正常的神經調節功能。
故若SMN1基因發生缺失等突變,雖有正常的 SMN2 基因,但整體產生的正常 SMN 蛋白仍不足以維持正常調節神經細胞的功能,進而成為SMA 患者。而患者中 SMN2 基因數目若越少,疾病程度將越嚴重。
依據SMN1基因分型,主要可分成以下三類:
| 基因分型 | 說明 |
| 正常人 | 一般人會擁有兩個以上的 SMN1 基因,運動神經元功能正常。 |
| 帶因者 | 擁有一個 SMN1 基因,外觀上與一般人無異,不影響正常發育,但有機率將此基因缺陷遺傳給下一代。 |
| 患者 | 完全沒有正常的 SMN1 基因,導致 SMN 蛋白濃度較低,影響運動神經功能正常運作。 |

▲SMN1 : SMN2基因比例將影響疾病嚴重程度。▲
脊髓性肌肉萎縮症(SMA)有不同的嚴重程度?
依照發病的年齡、嚴重程度與肌肉影響程度,可分成下列三型:
-
嚴重型:第一型重度脊髓肌肉萎縮症(Werdnig-Hoffmann Disease,SMA type I)
為最常見的SMA疾病分型。嬰兒在子宮內或出生後六個月內就會出現四肢及軀幹的肌功能減退、肌肉嚴重無力、哭聲無力等症狀,且通常無法控制頭部動作,也需要協助才可以坐立。第一型SMA患者胸廓大多呈現鐘型,以致出現呼吸與吞嚥困難的症狀,另外由於患者容易感染呼吸道疾病,因此通常三歲前就會因呼吸衰竭而死亡。
-
中間型:第二型中度脊髓性肌肉萎縮症(Dubowitz Disease,SMA type II)
症狀大多在出生後六個月至一歲半之間出現。第二型SMA患者可獨自坐立,但因為下肢呈對稱性之無力,導致通常無法自行站立或行走,偶爾可見患者舌頭及手部顫抖,肌腱反射消失或減弱,但臉部表情正常。由於持續性肌肉萎縮造成脊椎側彎,影響肺部正常功能,患者需終身仰賴支持性呼吸治療,除少數患者在孩童時期可能因併發呼吸道感染死亡,大多可以活至成年。 -
輕微型:第三型輕度脊髓性肌肉萎縮症(Kugelberg-Welander Disease,SMA type III)
發病年齡從一歲半至成年皆可能發生,這類型的患者可以自行站立或走動,但成年後近端肢體漸受影響,肌肉會愈發無力,且下肢較上肢嚴重,導致跑步、跳躍或上下樓梯會有輕度不便,仍經常需要倚靠拐杖走路或輪椅代步,不過此類患者長期存活率仍高。
脊髓性肌肉萎縮症是怎麼遺傳?
SMA 是透過體染色體隱性遺傳給下一代,若父母雙方同時為帶因者,會有 25% 機率懷上患有 SMA 的寶寶。由於患者的父母通常為帶因者,沒有明顯的家族史且不會表現出病徵,如沒有進行帶因者篩檢會不自知自己攜帶 SMN1 基因突變,進而忽略遺傳風險。
研究顯示國人 SMA 帶因率高達 1/50 1,即每50人約有1人會攜帶 SMN1 基因突變,屬於帶因率較高的遺傳性疾病,故建議夫妻在進行生育規劃時可先進行檢測自己是否為帶因者,評估遺傳機率。
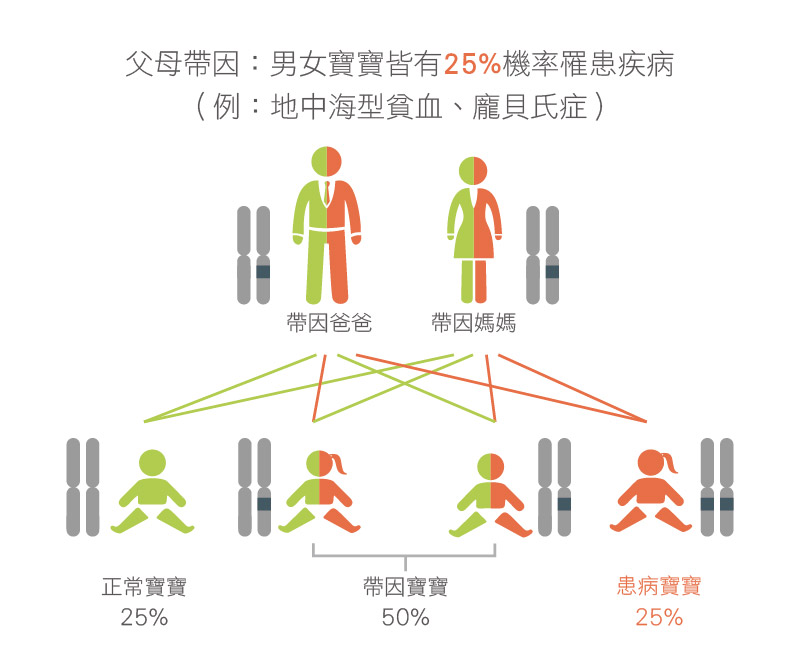
▲ 若夫妻雙方皆攜帶SMN1基因缺陷(帶因者),下一代將會有25% 的機率可能患上SMA。▲
脊髓性肌肉萎縮症(SMA)的治療與照護
目前SMA疾病尚無法治癒,但可依據患者不同的疾病分型與嚴重程度,借助藥物以延緩病程。目前美國 FDA 已批准三種治療 SMA 的藥物,主要針對 SMN1 及 SMN2 基因功能進行治療,藥價較為昂貴。在台灣已有兩項藥物(Spinraza、Zolgensma)通過藥物許可,自2020年7月1日起衛福部健保署已通過讓 Spinraza 納入健保給付。4
為減緩併發症的發生與提升生活品質,針對SMA患者的支持性治療也十分重要:
- 支持性呼吸治療:需減少呼吸系統的併發症,患者可能需要仰賴呼吸器及其他積極的支持性呼吸治療延長壽命。
- 物理性治療:以各種復健的方法來延緩脊柱側彎與髖關節脫臼,延長獨立行動的時間。
- 患者的營養照護:由於患者吞嚥及消化功能會受到影響,需要由專業營養師特別針對患者的營養狀況進行評估。
孕前或懷孕初期儘早做SMA基因篩檢,守護寶寶健康
由於SMA的基因突變導致的遺傳疾病成因於一般常規產檢無法檢測出來,目前臨床醫學上較安全的方式為夫妻懷孕前、懷孕早期可選擇進行夫妻雙方的「帶因者篩檢」,以瞭解雙方疾病的關鍵基因狀況,以評估遺傳給下一代的風險。
|
台灣基康提供單基因遺傳病的基因檢測,只需抽取受檢者的血液,進行基因分析,便可知道是否攜帶此基因缺陷,一生只需檢測一次。
|
檢測時機
婚前孕前、懷孕初期(建議 10~14 週前為佳)透過抽血則可進行SMA帶因者篩檢,以降低未來可能懷有患病寶寶的風險。
若於孕前檢測後,發現夫妻雙方帶有相同疾病基因缺陷,建議生育前可儘早安排與醫院遺傳醫師進行相關諮詢,向專業醫師了解夫妻間的狀況可能,或適時以胚胎著床前基因診斷(PGD/PGT-M)協助人工試管進行受孕,以降低胎兒異常風險;如為孕後發現,則建議可於懷孕 16~20 週左右時,進行羊膜穿刺以進行胎兒SMA之基因型確認情況。
檢測流程說明
一般可先透過聯繫檢測公司進行了解,安排於就近的配合診所內進行,流程為:
-
由專業人員為您說明檢測內容與衛教內容
-
填寫檢測同意書等資料
-
採集血液檢體約 5 ml
-
檢體於基因實驗室進行後續分析
-
抽血後等待報告書出具
-
由專業醫師為您提供報告說明與評估諮詢
參考資料
|
|
.png)
撰文/April Gan
文章最後更新時間:2022/04/18
(#台灣基康134)
.png)





